Table of Contents (click to expand)
A prion is a protein that changes its three-dimensional shape, which can cause disease. Prions are found in the brain and are resistant to proteases. The function of prions is not fully understood, but they are believed to play a role in intracellular signaling and cell adhesion. Prions are taken up by cells through receptors, and the process of formation is not fully understood.
Whenever we think of diseases or their causes, our mind often comes up with answers like bacteria and viruses. However, what if I told you that there is one particular miscreant that doesn’t have any nucleic acids or any cell organelles! It’s actually not even an organism. It’s a single biomolecule – Protein! Imagine that, nothing but a single molecule of protein that increases in number and can cause disease! But how can a protein replicate? Particularly without any enzymes or cellular machinery! Well, it doesn’t really replicate, but it does increase in number! Sounds crazy, right?
These crazy proteins are called Prions, a term coined by Dr. Stanley Prusiner. He and his team isolated the protein itself. Dr. Prusiner won a Nobel Prize in Medicine in 1997 for his research on prions. For a number of years, prions were nothing short of a mystery to researchers, as they were very difficult to isolate. Their functions are still not fully understood to date!
Prions are present all around your body, especially in the brain! So why are these same proteins causing problems? And if they’re present in everybody, why aren’t we all affected by their negative impacts? Short answer? The difference is in their structure! The prion protein in our body and the disease-causing prion proteins are different in terms of structure. Are you wondering what the structure has to do with function? Let’s take a quick look.
Recommended Video for you:
Protein Structure
Before we dive into prions, it’s necessary to understand the structure of a protein and how it relates to its function. Proteins actually have 4 levels of structure! Proteins are made from strings of amino acids, and these chains (also known as polypeptides) form their primary structure. Depending on these amino acids and the interaction via hydrogen bonding between them, two types of secondary structures can be formed, namely alpha-helix and beta-sheets. These structures then fold into three-dimensional structures. The folding is driven by hydrophobic interactions. The structure is kept intact via salt bridges (combinations of hydrogen bonds and ionic bonds) and disulfide bonds. Finally, multiple subunits come together and form a quaternary structure. This structure acts as a single unit and performs various functions.
This means that amino acid sequences determine the structure of the functional protein. Next is the connection between structure and function! Proteins do not randomly bind to their targets. They have a specific active site that is used to bind and continue its work. So, in the end, if the structure changes, it would change the shape of the active site. This would stop the protein from being able to do its job! Also, in certain cases, a change in structure would change the function and cause problems, as it might bind to targets not intended for that particular protein!
Now, time to delve into prions!
Structure Of Prions
Prion proteins (PrP) found in our body and those that cause diseases are structurally different. Some of them are even resistant to proteases (enzymes that degrade protein). We will look at both isoforms.
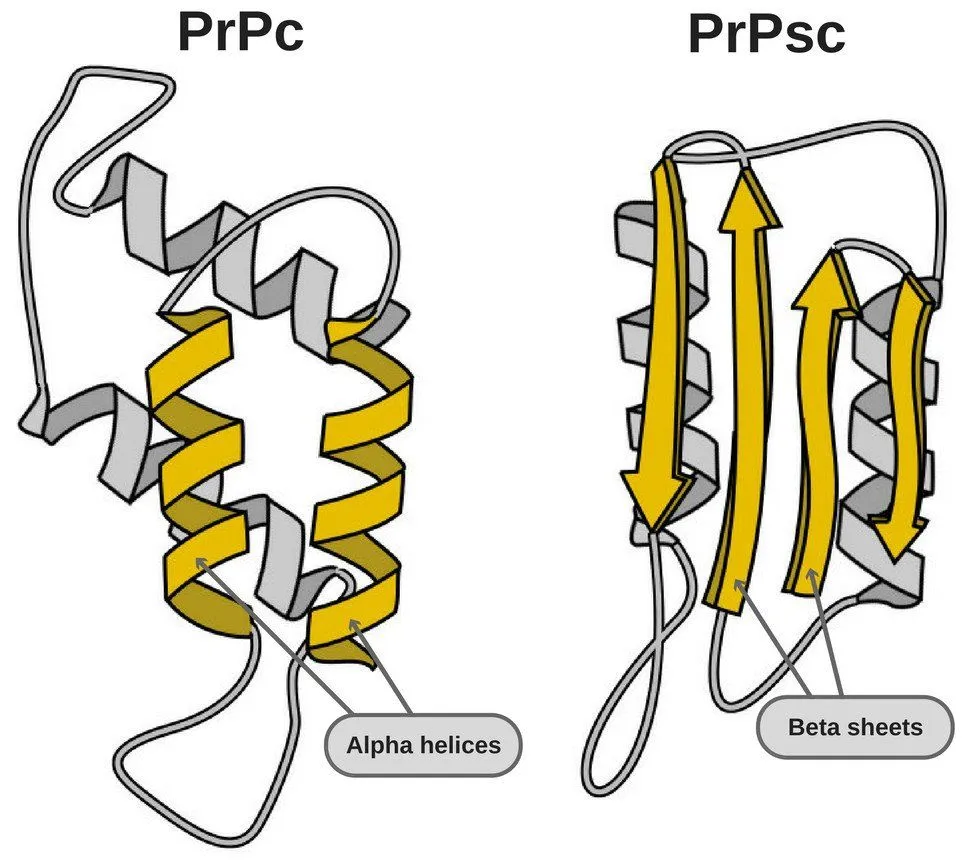
- PrPC – These proteins are found on the membranes of cells. They are believed to play important roles in intracellular signaling and cell adhesion. However, research is ongoing as the issues related to its function haven’t been solved.
- PrPSc – This disease-causing prion is protease-resistant. It changes the PrPC by affecting its conformation. The change in structure changes how it interacts and interconnects with proteins! The 3-D structure of these prions is not known, but what we do know is that they have more beta-sheets than the usual alpha-helix structure. This prion also forms highly structured amyloid fibers. The end of the fiber acts as a template for other free proteins to attach. Only similar prions with similar amino acids can bind! Cross-species binding is very rare, but it is possible.
Functions
Understanding the function of prions hasn’t been an easy task. Despite our technological advances, we haven’t gotten any clear proof of the functions it has in the body. The approach taken by our scientists was to splice the gene from the mice and look out for changes. Basically, this was to check, if we remove PrP, does anything go wrong or not? Such experiments have given unclear answers and the mice have shown only minor abnormalities.
A 2004 study showed that the lack of PrP genes in mice changed the long-term potentiation in the hippocampus. Also, another 2006 article indicated that PrP expression on stem cells is required for self-renewal of the bone marrow. The lack of PrP also made the cells sensitive to cell depletion.
How Do Prions Infect And Replicate?
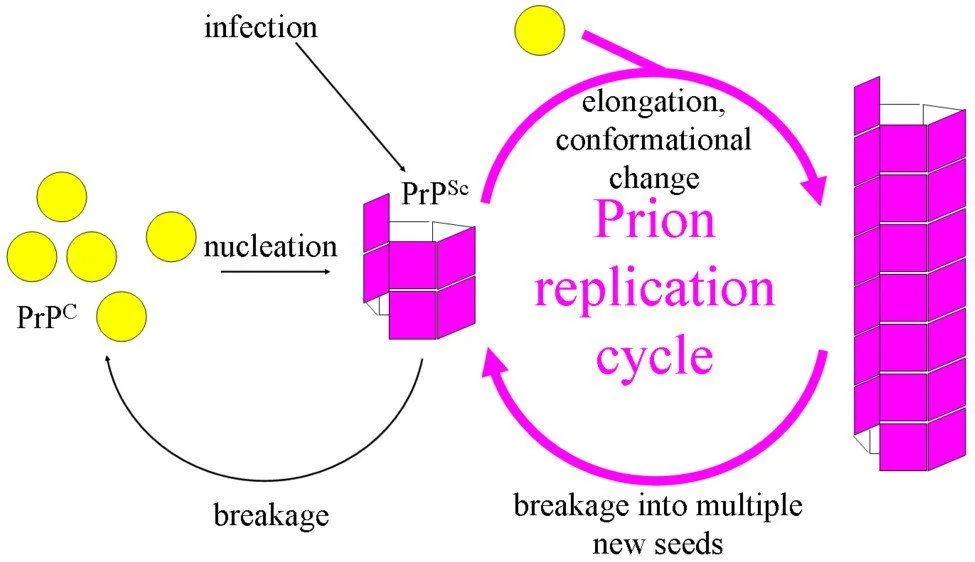
The topic of Prion infection and replication has been the most controversial one. Because, if you think about it, how can a protein molecule increase in number without using Transcription or Translation? Not having any cellular machinery should make the task of replication impossible. But prions do exactly that!
Similar to function, figuring out its replication mechanism hasn’t been an easy task. The infection of cells in vitro has been difficult and has mostly ended in failure. Most of the cell lines (that we use for experiments) that express the cellular PrP are resistant to prion infection. Even if certain cell lines are susceptible to a PrP strain, it will most likely be resistant to the other strains. Another problem faced by researchers is the lack of antibodies that can convincingly detect PrP(Sc). These problems combined have made the replication of prions difficult to understand.
Intake Of Prions
The initial step in understanding the entire process of infection is to know how prions are taken up by the cell. The rate of acceptance of prions by cell lines depends on how the preparations (containing prions) were made and if they were treated by an agent/chemical. Many studies have shown that strains known to be resistant to prions had taken up PrP(Sc). This made them think that receptors for PrP(Sc) are present in even the resistant cell lines! The receptors that have shown to affect the intake of prions into cells are LRP/LR, HS, and Proteoglycans. When knockout mice were used that lacked LRP/LR receptors, a drop in the intake of prions was observed. Such tests confirmed the fact that these receptors do play a key part.
Another method for the uptake of prions is macropinocytosis. A test was conducted where a macropinocytosis inhibitor was used to block the process and again, a fall in uptake was observed. These are just some of the known ways that uptake occurs.
Formation Of Prions
The process of formation is not fully understood, but researchers have been looking at various factors that affect its formation and could thus be playing a role in the formation. Studies have shown that it is a quick process and is initiated in a few minutes to a few hours. The PrP(Sc) production was often transient and did not end up as an infection. One of the things that are important for the formation of prions is lipid raft. It was observed that upon treatment by the drug filipin, the formation of prions stopped. The drug filipin depletes cholesterol and disrupts lipid rafts.
PrP(Sc) is mostly seen on the internal side with only a minute amount of them being on the cell surface. For the PrP(C) to be converted into PrP(Sc), it will need to be sent to the plasma membrane. Upon the removal of PrP(C), it was observed that PrP(Sc) accumulation reduced in the cell and in another experiment, the impaired transport of PrP(C) to the cell membrane effectively cured the cell, as it prevented PrP(Sc) formation. Even cells lacking GPI (Glycosylphosphatidylinositol) moiety were unable to sustain PrP(Sc) formation in the cell.
How Do Prions Spread From Cell To Cell?
There are two major ways of spreading prions into cells. The first way is via the release of PrP(Sc) into the cell culture medium and the second way is releasing them with exosomes. A study by Kanu and colleagues showed that cell-to-cell infection was reliant on cell proximity or direct contact. In some studies, PrP(Sc) was released into the culture, but transmission to nearby cells was preferred. The exact method of cell-to-cell infection is not well understood, but recent studies say that cytoplasmic bridges are involved. These bridges are known as tunneling tubes, formed of actin, that help in intercellular communication.
Prions Diseases
Prion diseases (collectively known as transmissible spongiform encephalopathies) comprise multiple conditions that can affect both humans and other animals. The most commonly known prion disease that affects humans is Creutzfeldt-Jakob Disease (CJD) and its variations. Some other known diseases are Scrapie (Sheep) and Bovine spongiform encephalopathies (commonly known as Mad Cow Disease).
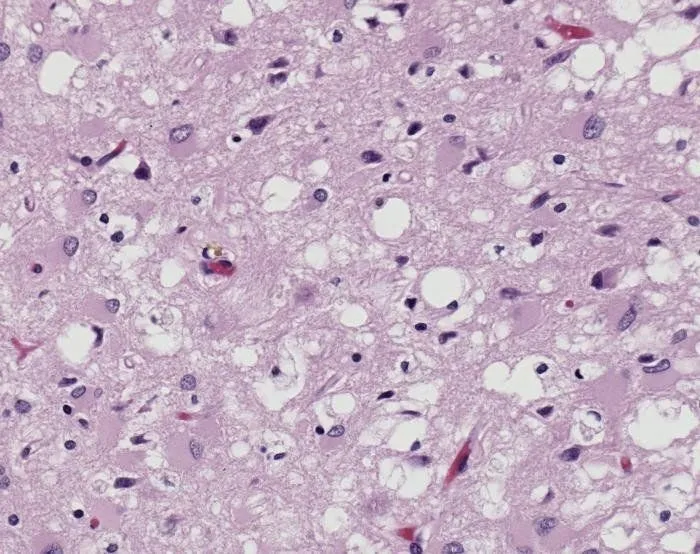
Prions cause neurodegenerative damage by accumulating outside the cells in the extracellular space. This happens in the central nervous system. Moreover, they come together to form amyloid fibers, which are toxic to the neurons and cause their death, leaving holes in the tissue, giving it a spongy look.
Studies have indicated that our body has two ways of degrading prions. First is lysosomal degradation and second is autophagy. So, why do prions still affect us? Because prions are smart and take out these mechanisms so that they can propagate! For example, Rab7 is a protein involved in lysosomal maturation, but it is observed that Rab7 is present in reduced levels in prion-affected cells. If Rab7 levels drop, it would affect lysosomal maturation. This means that fewer lysosomes will be present to degrade prions! Prions also affect the autophagy, which further helps them to advance and affect the neurons. These effects, in turn, affect our cells and help prions induce cell death. This induction of cell death results in the depolarization of the mitochondrial membrane, which quickens the process!
All in all, prions are by far the most mysterious agents that cause diseases. With a lot still unknown (e.g., functions, pathophysiology and many others), it will be exciting to wait and see what further research digs up.
References (click to expand)
- http://web.archive.org/web/20190904000207/http://www.learner.org:80/courses/biology/textbook/proteo/proteo_4.html
- Pan, K. M., Baldwin, M., Nguyen, J., Gasset, M., Serban, A., Groth, D., … Cohen, F. E. (1993, December). Conversion of alpha-helices into beta-sheets features in the formation of the scrapie prion proteins. Proceedings of the National Academy of Sciences. Proceedings of the National Academy of Sciences.
- Grassmann, A., Wolf, H., Hofmann, J., Graham, J., & Vorberg, I. (2013, January 22). Cellular Aspects of Prion Replication In Vitro. Viruses. MDPI AG.
- Yao, H., Zhao, D., Khan, S. H., & Yang, L. (2013, June 1). Role of autophagy in prion protein-induced neurodegenerative diseases. Acta Biochimica et Biophysica Sinica. China Science Publishing & Media Ltd.
- Shim, S. Y., Karri, S., Law, S., Schatzl, H. M., & Gilch, S. (2016, February 11). Prion infection impairs lysosomal degradation capacity by interfering with rab7 membrane attachment in neuronal cells. Scientific Reports. Springer Science and Business Media LLC.