The “Patient Zero” of a viral pandemic is the first individual to be infected by the disease. Scientists attempt to locate this patient using a host of genetic, evolutionary and epidemiological models.
The word ‘pandemic’ has become a household term in the past 18 months, and rightfully so, but what exactly is a pandemic?
A pandemic is the spread of a disease to different countries and continents that causes widespread infections in a large population.
Our real-time experience of a pandemic may seem novel, but in reality, pandemics have occurred quite regularly in the history of human existence. A few of these historical pandemics include the bubonic plague of the mid-1300s, the cholera pandemic of the 19th century, the 1918 Spanish Flu, and the pandemics of the 21st century caused by viruses such as H1N1, H2N2, and the coronaviruses (SARS and MERS).
Scientists have worked tirelessly to investigate the causes and effects of these viruses in order to treat the infections caused by them. One such vital piece of information about these viruses is tracing the first case of their origin.
Recommended Video for you:
Why Is It Important To Trace The First Case Of An Outbreak?
We’ve all asked this question at least once: “Where does the virus come from?” Aside from curiosity, tracing the first case of a viral outbreak provides scientists with information about several factors that ultimately help mitigate the spread of the virus (as represented in movies such as Outbreak and Contagion). These factors include:
- Figuring out the origin of the infection.
- Providing information about how further outbreaks of a similar nature can be prevented.
- Preventing the immediate spread of the disease (this is important for infections occurring in a relatively small area, such as a building).
However, before diving into how such first cases are traced, we should familiarize ourselves with a few related terms.
In zombie apocalypse films, the first zombie the authorities encounter is never the first person to become a zombie. In this case, the first person to be diagnosed by scientists as “infected” is called an index case, while the first person to trigger the zombie apocalypse is known as a primary case.
The first person to be infected is sometimes referred to as “patient zero“. This term surged into popularity during the HIV epidemic due to a translation error. The first person to officially have an HIV infection was depicted on paper as “Patient o”, which was mistakenly read as “Patient Zero”.
Since the outbreak of the Covid-19 pandemic, we have often come across the hypothesis that the origin of the SARS-CoV-2 infection was traced to the Huanan seafood market in Wuhan, China. This may well have been the index case, but scientists believe that it is not the primary case. The search for the primary case of the Covid-19 virus is ongoing.
So, How Is The First Case Of A Viral Pandemic Traced?
One of the first tasks for scientists in the event of a viral epidemic is to search for the primary case. This is done according to the type of pathogens that cause the disease.
Zoonotic diseases are a type of viral disease caused by pathogens that jump from animal to human hosts. This jump from an animal to a human host can occur for various reasons, including climate change, food habits of humans, and humans encroaching upon natural habitats; anything that increases the chances of humans interacting with wild animals. Some examples of zoonotic diseases include SARS and MERS (coronavirus), swine flu, Ebola, avian influenza, Zika virus, Nipah virus, and HIV.
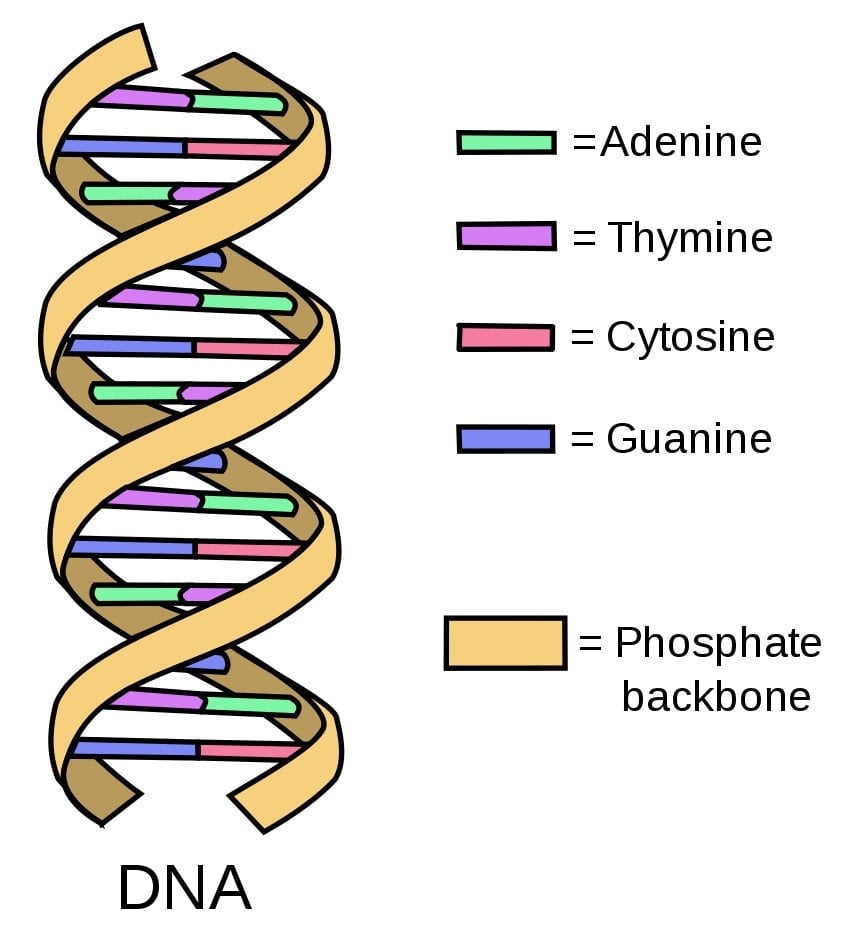
Scientists use DNA comparison to determine how an animal host is able to infect humans with a particular virus.
DNA is composed of small building blocks known as nucleotides. Think of nucleotides as Lego pieces that come in four kinds—A (adenine), C (cytosine), T (thymine), and G (guanine)—stacked on top of each other randomly to form DNA. These four nucleotides differ in their arrangement in all organisms, including humans, animals, and even viruses.
Scientists isolate the pathogen (an organism that causes disease) from humans and animals and compare its DNA. The more similar the arrangement of the nucleotides, the more closely related the DNA sequences are. The closeness of the DNA sequences (defined by the sameness of the arrangement of nucleotides of the DNA) tells the scientists which animal host the virus used in its jump to humans.
For the SARS-CoV-2 coronavirus, the closest match was found to be a virus in bats (a 96% match). However, knowing the probable animal host of a virus alone does not provide answers about the primary case, since animals are more likely to interact with other animals than with humans.
Think of a bat (the host of most of the viral pathogens mentioned above) that is infected by a virus and nibbling on some fruit, which then drops on the street. Another animal, like a cat, comes across the fruit and curiously licks it. The cat then goes on to interact with humans, thus exposing them to the virus. Here, the cat becomes an intermediate host animal.
This chain of events makes it even more difficult for scientists to trace the origin of a viral infection. In the case of Covid-19, scientists believe this to be the case, since the percentage of closeness between the bat and human coronaviruses is not high enough.
With each jump, from the host animal to the intermediate animal and then eventually to humans, the virus undergoes random mutations. These mutations cause a change in the virus structure, such as the formation of a new type of spike on the coronavirus each time it jumps from one organism to another. This new spike allows it to infect an organism aside from the bat. With the formation of a new spike, the arrangement of the nucleotides of the virus changes as well!
Hence, even if it’s the same virus jumping from a bat to a cat, and then on to humans, there are differences in the arrangement of nucleotides in its DNA.

Scientists need to keep track of the path of the virus as it jumps among all these animals. They do this by forming a family tree.
Scientists place the virus isolated from bats at the position of the grandparents at the top of the tree. Next, they position the mutated virus that jumped into the cat below the grandparents, where the parents would normally be placed. And then, finally, the last position in the family tree beneath the parents (the space for the children) is taken by the virus that infects us, humans.
The above is a simplified representation of how such a tree would look. In reality, they are much more complex, consisting of numerous viruses branching out from the one isolated from the primary host.
This kind of family tree connecting the relatedness of DNA is known as a phylogenetic tree. They help scientists understand the degree of closeness between the viruses present in different organisms.
Finding The Primary Case Is Still Difficult.
Even after scientists have narrowed down the animal sources of a pathogen, it is still difficult to figure out the primary case of the disease. A few reasons for that are:
- It is difficult to sample the virus from wild animals both due to safety concerns and the danger of the scientists getting infected.
- Even if scientists narrow down on the particular species of animals from which the virus jumped into humans, the rate of viral infection in humans is very high. Hence, instead of only one person being sick at first, there must have been a group of people who showed symptoms, which makes identifying patient zero difficult.
- A virus mutates very rapidly (like forming new spikes that change the arrangement of their nucleotides), even when transmitted from human to human, making it increasingly difficult to trace its origin.
Scientists are still working on finding the primary cause of not only the SARS-CoV-2 virus, but also other viruses that continue to ravage certain parts of the world.
References (click to expand)
- Dictionary of Epidemiology, 5th Edition - Academia.dk. academia.dk
- Bonneaud, C., & Longdon, B. (2020, August 30). Emerging pathogen evolution. EMBO reports. EMBO.
- How virus detectives trace the origins of an outbreak. The Conversation
- McKay, R. A. (2014). “Patient Zero”: The Absence of a Patient’s View of the Early North American AIDS Epidemic. Bulletin of the History of Medicine. Project Muse.
- Grubaugh, N. D., Ladner, J. T., Lemey, P., Pybus, O. G., Rambaut, A., Holmes, E. C., & Andersen, K. G. (2018, December 13). Tracking virus outbreaks in the twenty-first century. Nature Microbiology. Springer Science and Business Media LLC.
- Zoonotic Diseases | One Health | CDC. The Centers for Disease Control and Prevention
- Zhou, P., Yang, X.-L., Wang, X.-G., Hu, B., Zhang, L., Zhang, W., … Shi, Z.-L. (2020, February 3). A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature. Springer Science and Business Media LLC.