Table of Contents (click to expand)
Gel electrophoresis is a go-to technique for the charge- and size-based separation of nucleic acids in biology. This separation forms the basis of several biological experiments.
We often read about scientists working with DNA, extracting DNA from different samples, manipulating them, joining them, cutting them into smaller pieces, using enzymes and so on. The prerequisite for all these techniques is to be able to accurately visualize the DNA.
Have you ever wondered how scientists are able to visualize the tiny DNA molecule? Well, the answer is “electrophoresis”.
This is a method to separate charged biomolecules like DNA, RNA and proteins using electricity. Arne Tiselius used electrophoresis to separate biological molecules for the first time in 1931. Even though it’s been almost a century, the technique of electrophoresis is still an important technique and the only way to separate complex biomolecules.
Recommended Video for you:
What Is The Principle Behind This Technique?
The main principle behind “electrophoresis” is using electricity to separate biomolecules based on their charge and size. The separation is based on the simple fact—”Like charges repel each other and opposite charges attract each other”.
Most biomolecules are charged, either positively or negatively. DNA and RNA have a negative charge due to the phosphate groups present in their structure, while proteins can be either positive or negative, depending on the amino acids that make them up.
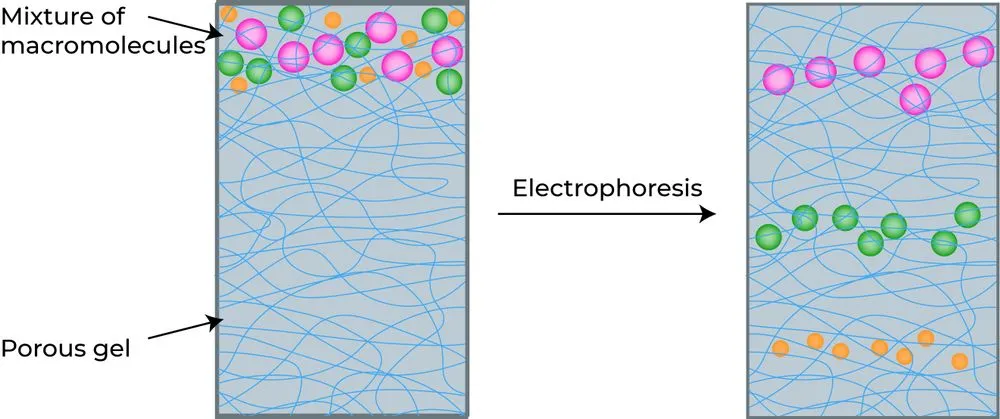
If this separation is done on a jelly medium, usually made of agarose (a carbohydrate polymer), it is called agarose gel electrophoresis.
What Is The Gel In Gel Electrophoresis?
The gel in the gel electrophoresis refers to the matrix on which the biomolecules are separated. It is clear and jelly-like in nature, like the gelatin we consume as food.
The most commonly used matrix for electrophoresis is the polysaccharide agarose. You may wonder why agarose is used in this technique, and there are two reasons. One is its ability to form a mesh (crosslink). The agarose matrix acts as a sieve or mesh, through which the biomolecules can easily travel, depending on their size. The second reason is its neutral nature, which ensures that there is no reaction between agarose and the biomolecules to be separated, thus minimizing the chances of false results.
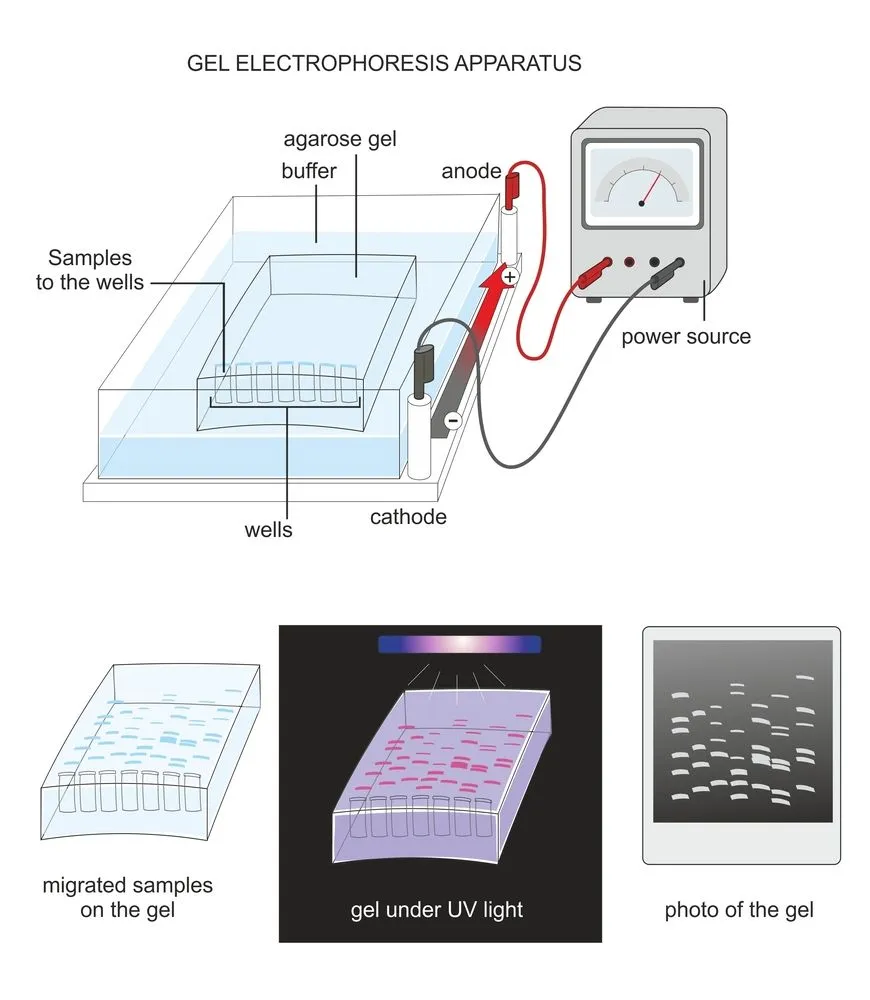
Let’s look at the setup of gel electrophoresis to understand how the separation takes place.
The biomolecules move through the gel based on their charge and size. Negatively charged biomolecules move towards the positive terminal and vice versa.
The smaller fragments often have low molecular weight, meaning that they are lighter. This enables them to travel swiftly through the gel, covering a larger distance. On the contrary, the larger fragments have a higher molecular weight, so they move slowly through the gel, as the pore size of the agarose will not let the larger molecules move faster.
The movement is also based on the percentage of agarose used in the gel. Low-percentage agarose gels have larger pore sizes to facilitate the rapid movement of larger molecules. For the separation of smaller DNA molecules, a higher percentage of agarose gel is better, as the smaller pore size can provide better separation.
How To Visualize The DNA
If we look at the electrophoresis buffer, the agarose gel or the DNA samples, they are all transparent. So, how do we visualize the DNA? We use colored dye that binds to the DNA or the biomolecules. The most commonly used DNA binding dye is ethidium bromide (EtBr) .
The dye is either added to the agarose gel or to the buffer filled in the apparatus. Either way, it should come in contact with the DNA and bind to it.
With this dye added, the gel is exposed to ultraviolet (UV) light. Under UV light, ethidium bromide releases a bright light. As ethidium is already bound to the DNA, the places on the gel that appear bright are the places where the DNA can be found.
One should always take precautions while handling EtBr, as it is cancer-causing (carcinogenic). It must be used with caution and disposed of properly.
The DNA will be seen as bright small vertical lines, called bands, against a dark background. Each band consists of DNA molecules of the same size. Intact bands indicate good-quality DNA. If the DNA is of low quality or has contaminants, the DNA band will not be intact, but spread out, called a smear.
If we want to determine the size or molecular weight of the DNA, we can use DNA ladders, which are DNA samples of known sizes that are available commercially. If we load the DNA ladder along with the sample DNA (whose molecular weight is unknown), the distance migrated by both the DNA samples can be compared to determine the molecular weight of the sample DNA.
What Factors Can Influence DNA Migration In The Gel?
Several factors will influence the way DNA may migrate within the gel. There is an inverse relationship between the size of the DNA molecule and the concentration of agarose used; larger DNA requires smaller concentrations of agarose and vice versa.
It also depends on the voltage applied. The samples move faster under high voltage, but the DNA bands will not remain intact. A lower voltage will result in the slower migration of DNA, but the bands will be distinct. Therefore, one may have to compromise between the time and voltage for proper DNA separation.
The presence of EtBr, the quality of agarose, and the pH and composition of the buffer employed also influence DNA mobility.
A Final Word
Agarose gel electrophoresis is the key, and probably the only technique to determine the quality and size of nucleic acids in several fields of biological research. The last two decades have seen a sweeping period of evolution and development in the technique, as well as an expansion of the instances where it may be used.
References (click to expand)
- Electrophoresis - an overview | ScienceDirect Topics.
- Agarose Gel Electrophoresis - an overview.
- Lee, P. Y., Costumbrado, J., Hsu, C.-Y., & Kim, Y. H. (2012, April 20). Agarose Gel Electrophoresis for the Separation of DNA Fragments. Journal of Visualized Experiments. MyJove Corporation.
- Motohashi, K. (2019, September 9). Development of highly sensitive and low-cost DNA agarose gel electrophoresis detection systems, and evaluation of non-mutagenic and loading dye-type DNA-staining reagents. (R. Kalendar, Ed.), Plos One. Public Library of Science (PLoS).
- Sigmon, J., & Larcom, L. L. (1996, January). The effect of ethidium bromide on mobility of DNA fragments in agarose gel electrophoresis. Electrophoresis. Wiley.