A Polymerase Chain Reaction or PCR is a method to create thousands of copies of a DNA strand. It exploits the ability of the polymerase enzymes to create copies of the genetic material under laboratory conditions.
DNA (deoxyribonucleic acid) forms the foundation for countless research studies involving living organisms. From the DNA code, we can glean the genetic basis of diseases, develop drugs, perform forensic testing, identify microbes, and so much more.
The most basic thing needed for such research is large quantities of the DNA fragment under investigation. However, DNA isolated from the cells, tissues or any other biological source is often not enough for the analysis. Thus, scientists need to make more copies of DNA.
This is where the crucial role of the “Polymerase Chain Reaction” comes into the picture.
Recommended Video for you:
What Is A Polymerase Chain Reaction?
PCR exploits the ability of the polymerase enzymes to create copies of the genetic material under laboratory conditions.
Before PCR, copies of DNA were made by isolating a particular DNA fragment and inserting it into the genome of living cells. The living cells replicated the inserted DNA while replicating their own DNA. The technique was a laborious and time-consuming way to generate DNA copies sufficient for further study.
However, this is not the case any longer. Most of the credit for this advancement goes to Kary Mullis, who invented “Polymerase chain reaction” (PCR) in 1983, marking the beginning of the “Biotechnology Revolution”. Today, PCR is a very common laboratory technique even in smaller labs, and is used to generate DNA copies on a regular basis.
PCR can selectively make copies of the DNA of interest through a process often known as “molecular photocopying”. Once several copies of the DNA are synthesized using PCR, the DNA is then “amplified”.
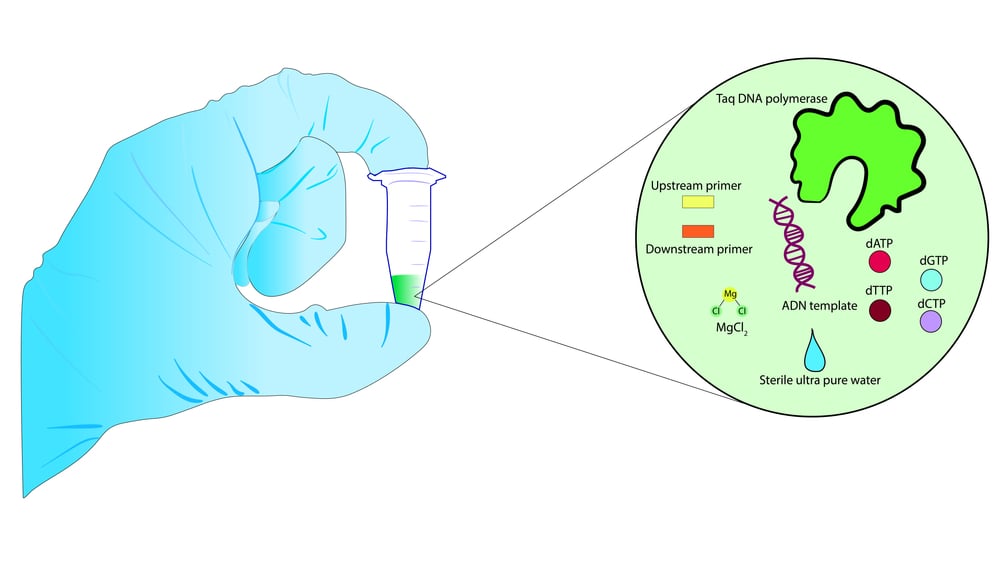
What Are The Components Of A PCR Reaction?
The key components of a PCR reaction are the template DNA, primers, nucleotides, and heat-stable DNA polymerase. Let’s briefly learn about each of these components.
DNA from the simplest bacteria to the most complex animals and plants can be used for PCR. However, the whole DNA (template DNA) isn’t put through the PCR; only a small section will be amplified during the process.
For the DNA to be amplified, primers—short stretches of nucleotides (approximately 20 bp)—are very important. A set of primers are used, both a forward primer and a reverse primer. The primers bind to the start and end of the DNA strands, signaling the points from which the DNA strand needs to be amplified.
Nucleotides used for the PCR reaction are a mixture of all four nitrogenous bases found in DNA. They are adenine (A), thymine (T), guanine (G), and cytosine (C).
The last and most important component is DNA polymerase. A polymerase enzyme makes new DNA molecules by assembling the nucleotides, in a manner that is complementary to the existing strand. In this way, it can actually create two identical DNA molecules from a single strand of DNA.
Along with the enzyme, the cofactors required for the action of DNA polymerase must be added to the reaction mixture. Co-factors are the metallic compounds that are critical to enzyme activity. Magnesium ions are the cofactors for DNA polymerase.
The DNA polymerase used in PCR is the thermostable DNA polymerase (often called Taq polymerase) isolated from thermophilic organisms that can survive high temperatures.
What Are The Steps Of A PCR Reaction?
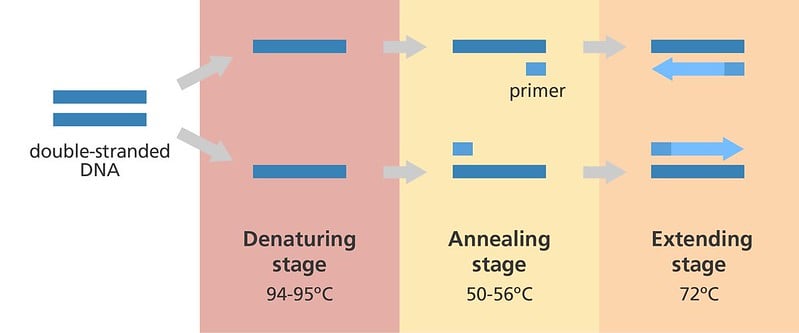
Now that we know all the components required for PCR, let’s check out the three steps involved in a PCR reaction. The steps are denaturation, annealing, and extension.
The activity of the polymerase is dependent on the presence of single single-stranded DNA to which the primers can bind. This can simply be achieved by heating the DNA sample at 94-98°C.
Heating breaks the bonds holding the two DNA strands together. The process is called denaturation, as the double-stranded DNA molecule is denatured to two single-stranded molecules. One DNA strand will be called the template strand, while its pair is called the complementary strand.
The next step requires the primers to bind (anneal) to the template DNA at specific sites. The forward primer binds to the start of the template DNA (one strand on the double-stranded DNA) at the 3 bp nucleotide sequence ATG (start codon). The reverse primer binds to the end of the complementary DNA (the second strand of the double stranded DNA) strand at the 3 bp nucleotide sequences TAG, TAA or TGA (stop codons). The DNA between the start and stop codons gets amplified.
The success of this step depends on the primer sequence and the temperature chosen for annealing, typically 50-65°C.
The last and final step is elongation or termination, which occurs at 72°C, the optimum temperature for the activity of Taq polymerase. The DNA polymerase will recognize the primer-bound region of DNA and add nucleotides complementary to the template DNA strand. This occurs until it encounters the second primer.
After a successful termination reaction, there will be two DNA helices instead of the one DNA helix used in the initial stage. In each of the two DNA helices, one strand will be the original strand provided by the DNA sample. The other strand will be the complementary strand synthesized by the DNA polymerase during PCR.
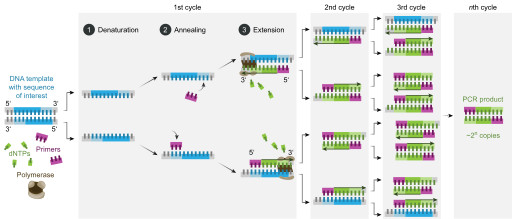
What Is The Principle Behind DNA Amplification Using PCR?
The steps of denaturation, annealing, and polymerization comprise one PCR cycle. A typical PCR reaction may require 25-35 cycles for optimal DNA amplification.
At the end of one cycle, the single DNA template will form two DNA molecules. At the end of two cycles, the two DNA will form four DNA molecules that will then amplify to eight DNA molecules at the end of three cycles. At the end of n cycles, there will be 2n copies of the original DNA template.
After each cycle, the number of DNA molecules that can act as templates for the next cycle increases exponentially. This increase in the template number cycle after cycle is the basis behind the amplification of DNA molecules in PCR.
What Are The Pros And Cons Of PCR?
The key pros of the PCR technique are that it is capable of producing millions to billions of copies of DNA in a matter of just a few hours. The technique is rapid, relatively easy to learn, and can be performed under basic laboratory conditions.
The major disadvantage is that the technique is highly sensitive, so the sample used for amplification must be free of contaminants. Even small traces of unwanted DNA may get amplified along with the DNA of interest, giving false results.
Another drawback is the requirement of sequence information of the DNA to design primers. Additionally, the primers may occasionally anneal to the wrong sites of DNA. Such non-specific annealing of the primers may result in the amplification of the wrong fragment of DNA.
In a rare scenario, DNA polymerase may incorporate a wrong base, leading to a change in the sequence of the DNA of interest, which may affect the downstream process.
For a successful PCR reaction, all you need to have is pure genetic material, an appropriate set of primers, and a proper annealing temperature.
Where Can This Technology Be Used?
The primary application of PCR is selective DNA isolation from mixed DNA samples. This can help diagnose infectious diseases, genetic diseases, and several types of cancers in a short period of time. It is also used in forensic sciences to identify criminals and test for parentage.
It forms the basis of most applications involving molecular biology, gene cloning, recombinant DNA technology, and mutagenesis.
Conclusion
The practical advancement of the simple and versatile PCR technique, as prescribed by Kary Mullis, has drastically transformed biological research. All we have to do is mix all the components of PCR in the appropriate concentrations in a small tube, load it into the PCR machine (thermocycler) and wait a few hours. After the waiting period, researchers will have millions to a billion copies of your DNA of interest. Isn’t that amazing?
This is why PCR will always be a rapid and reliable technique for generating DNA copies, as compared to the methods used prior to this game-changing discovery.
References (click to expand)
- Garibyan, L., & Avashia, N. (2013, March). Polymerase Chain Reaction. Journal of Investigative Dermatology. Elsevier BV.
- PCR and Cloning Expressed Genes | Learn Science at ....
- Valones, M. A. A., Guimarães, R. L., Brandão, L. A. C., Souza, P. R. E. de ., Carvalho, A. de A. T., & Crovela, S. (2009, March). Principles and applications of polymerase chain reaction in medical diagnostic fields: a review. Brazilian Journal of Microbiology. FapUNIFESP (SciELO).
- The applications of the polymerase chain reaction in ....
- Powledge, T. M. (2004, June). The polymerase chain reaction. Advances in Physiology Education. American Physiological Society.